Validation des logiciels pour les systèmes qualité des dispositifs médicaux
Maurice Navarro
Consultant qualité & logiciel pour dispositifs médicaux
La validation des logiciels pour les systèmes qualité des dispositifs médicaux est requise à trois niveaux de la norme ISO 13485 :
- les exigences générales sur le système de management de la qualité,
- les exigences de validation des processus de production et de prestation de services et
- les exigences de maîtrise des équipements de surveillance et de mesure.
A quoi correspondent ces exigences ? Comment les mettre en œuvre efficacement ? Quel est l'intérêt pour le fabricant de dispositifs médicaux ?
Traitons ces questions à l'occasion de cet article.

Qu'est-ce que la validation des logiciels pour les systèmes qualité des dispositifs médicaux ?
L'un des objectifs d'un système de management de la qualité (SMQ) ISO 13485 est d'identifier et de maîtriser les évènements potentiels susceptibles d'impacter la qualité des dispositifs médicaux. L'objectif est de réduire leur probabilité d'occurrence (leur risque).
Parmi ces évènements potentiels figure le dysfonctionnement des logiciels utilisés par l'entreprise.
En effet, directement ou indirectement, le dysfonctionnement d'un logiciel peut amener la mise sur le marché de dispositifs médicaux non conformes à leurs spécifications.
Sur cette base, valider les logiciels pour les systèmes qualité des dispositifs médicaux revient à confirmer que les logiciels pouvant impacter la sécurité ou la qualité des dispositifs médicaux fonctionnent correctement au regard de leur finalité d'utilisation.
Cette validation doit être réalisée avant la première utilisation du logiciel considéré, après ses mises à jour et suite à leur extension ou modification d'utilisation.
Ces applications logicielles doivent être validées avant leur première utilisation et, lorsque approprié, après la modification de ce logiciel ou de son application.
Source : ISO 13485
Comment mettre efficacement en œuvre cette exigence ?
Le rapport technique ISO/TR 80002-2 (Logiciels de dispositifs médicaux — Partie 2 : Validation des logiciels pour les systèmes de qualité des dispositifs médicaux) est un guide de validation des logiciels pour les systèmes qualité des dispositifs médicaux.
Dans le cadre d'une entreprise on distingue deux grands types de logiciels :
- les logiciels achetés à des fournisseurs tiers et
- les logiciels développés en interne.
L'ISO/TR 80002-2 est applicable à ces deux types de logiciels pour systèmes de management de la qualité ISO 13485.
En ce qui concerne les logiciels développés en interne, ce rapport technique invite à une maîtrise de conception des logiciels qui, partant de l'analyse de risques des processus, assure leur validation.
Quant aux logiciels achetés à des fournisseurs tiers, la validation doit être mise en œuvre sur la base d'un plan de validation pré-établi.
Dans ces deux cas de figure, mettre efficacement en œuvre la validation du logiciel revient à déterminer l'étendue des essais de validation auxquels il doit être soumis.
Le rapport technique ISO/TR 80002-2 indique que l'élément de pondération est l'analyse de risques des processus qui utilisent ces logiciels.
En effet, l'analyse de risques des processus vous permet d'adapter l'étendue des essais de validation pour mettre efficacement en œuvre les exigences de la norme ISO 13485.
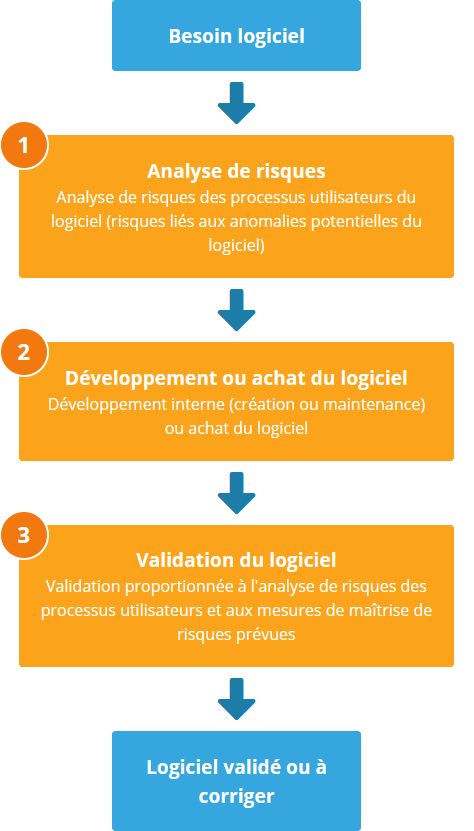
Note : tout logiciel présentant des risques, directs ou indirects, pour la sécurité ou la sureté des dispositifs médicaux doit être validé quel que soit son niveau de risque.
Intérêt pour les fabricants de dispositifs médicaux
Les logiciels développés en interne ont souvent pour objectif de répondre à des besoins spécifiques de l'entreprise tels que des réglages particuliers à réaliser lors de la production des dispositifs.
Dans ce contexte, la maîtrise de conception et la validation des logiciels présente un intérêt important : se protéger contre les conséquences, notamment les surcoûts, que pourrait générer un dysfonctionnement du logiciel lors de son utilisation.
Par contre, la validation des logiciels achetés à des fournisseurs tiers se prête à discussion.
En effet, le client d'un fournisseur de logiciel peut légitimement s'attendre à ce que le logiciel qu'il a acheté ait fait l'objet d'une validation et fonctionne correctement.
Ce n'est malheureusement pas toujours le cas. De ce fait, par la validation du logiciel, le fabricant se protège aussi contre les dysfonctionnements de ces logiciels lors de leur utilisation.
Pour en savoir plus
Il est important de faire la différence entre le développement d'un logiciel (de) dispositif médical et le développement d'un logiciel pour le système de management de la qualité (SMQ).
Le développement de logiciels pour le système de management de la qualité (SMQ) requiert un processus de génie logiciel identifié.